2017-07-21 | 免疫细胞及胶质细胞调控突触可塑性及慢性痛/Immune and glial regulation of neural plasticity and chronic pain
报告人:Dr. Ru-Rong Ji
整理人:田纳西
审核人:王韵
2017年7月21日下午,来自杜克大学 (University of Duke) 痛觉研究中心主任Ru-Rong Ji(纪如荣)教授受IDG/麦戈文脑研究所王韵教授的邀请来到北京大学,在王克桢楼1113会议室为大家带来了一场题为“Immune and glial regulation of neural plasticity and chronic pain”的学术报告。纪如荣教授从事神经科学和痛觉研究20余年,率先在国际上开展蛋白激酶MAPK调节慢性痛的神经可塑性研究,也是国际上研究非神经元和胶质细胞调节疼痛的先驱者之一。本场讲座中,纪如荣教授综述了近20年来慢性痛研究进展,并介绍了实验室最新的研究成果,即癌症细胞分泌的PD-L1具有镇痛效果。
慢性痛研究回顾
疼痛是由现实或潜在的组织损伤引起的不愉快的感觉性和情绪性体验。痛觉的产生通过上行传入系统实现:外周伤害性感受器受到内源或外源伤害性刺激产生神经冲动传至胞体即背根神经节进行整合,再经由低级中枢脊髓背角向丘脑传入整合,投射至躯体感觉皮层产生痛觉的感觉成分。
慢性痛的产生包括外周敏化和中枢敏化。在外周,各类致炎介质作用于神经末梢感受器上的受体,通过信号转导、蛋白激酶等调控方式改变离子通道的表达及功能,使细胞兴奋性升高,对伤害性刺激反应性增强,造成外周敏化(图1)。中枢敏化的观点是1983年由Clifford J. Woolf教授提出,这之后的研究也陆续证实,在脊髓和相关脑区均可发生突触可塑性及神经环路重塑等长时程变化。

图1
在脊髓背角,兴奋性神经元的功能上调和抑制性神经元的功能下调可放大和维持痛信号的传入。此外,星形胶质细胞和小胶质细胞等非神经元成分也可能参与慢性痛形成。纪如荣教授2013年发表在Pain上的一篇综述中提出,慢性痛是由胶质细胞功能失调引起的。脊神经损伤3至7天后,脊髓可见CD11b阳性(小胶质细胞标记物)细胞数明显上调,即小胶质细胞大量激活,同时表达p-p38、产生和分泌白细胞介素TNF-α和IL-1β。在神经损伤后2周以后,脊髓小胶质细胞激活逐渐下调,而脊髓星形胶质细胞则产生了长时程激活。在坐骨神经损伤3周时,可在脊髓观察到大量GFAP阳性(星形胶质细胞标记物)细胞,同时表达Cx43,增强星形胶质细胞间的胞间联系并产生和分泌趋化因子CCL2和CXCL1等。胶质细胞分泌的细胞因子如TNF-α作用于神经元可直接上调NMDA介导的电流、产生长时程增强(long term potentiation, LTP),增加兴奋性突触的传递;IL-1β则可下调GABA和Glycine介导的抑制性电流,产生去抑制的作用。
纪如荣教授2016年发表在Science的综述中总结了胶质细胞调节慢性痛的方式。胶质细胞本身虽然不能传导疼痛,但是可以通过与神经元的相互作用调节痛信号传递,在慢性痛中发挥放大器的作用。首先,初级感觉神经元的中枢突末梢释放趋化因子激活小胶质细胞,小胶质继而释放TNF-α、IL-1β和BDNF等因子调节突触后神经元的兴奋性,同时激活星形胶质细胞释放趋化因子及递质等,共同调节突触后神经元;此外,小胶质与星形胶质细胞释放的细胞因子也可作用于突触前的感觉神经元末梢,增加疼痛的敏感性和持续时间,使急性痛发展为慢性痛(图2)。
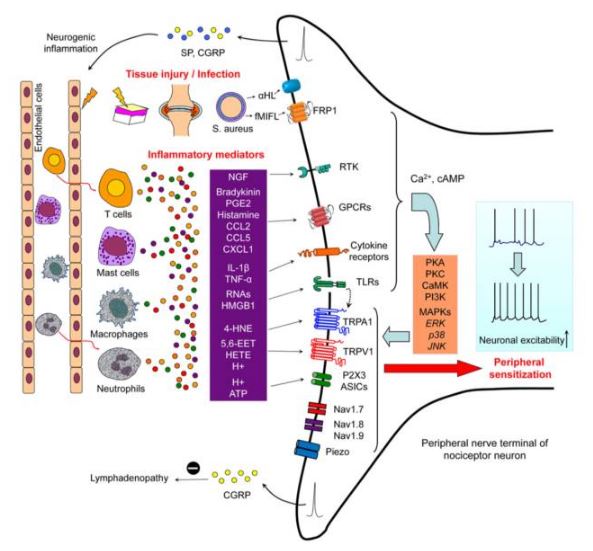
图2
神经炎症(Neuroinflammation)是发生在外周和中枢神经系统的炎症。临床上认为神经炎症可以引起各种精神疾病,如阿尔兹海默症、帕金森和抑郁症等。神经炎症的发生包括免疫细胞浸润、胶质细胞激活和产生细胞因子及趋化因子,其中小胶质和星形胶质细胞的激活为神经炎症的主要标志。基于以上研究内容及神经炎症的特性,纪如荣教授提出了神经炎症可以驱动中枢敏化和慢性疼痛这一理论。
肿瘤与疼痛简介
初级感觉神经元除了可受胶质细胞调节外,也可以和免疫细胞及肿瘤细胞等发生相互作用,根据非神经元产生的介质类型不同,产生致痛或是镇痛的效果(图3)。临床上癌症痛是癌症的常见症状。一方面来说,肿瘤细胞可以产生炎性介质激活神经末梢感受器,上调神经元的敏感性产生痛敏;另一方面,NGF和VEGF等生长因子介导的通路可使肿瘤中伤害性感受神经末梢的支配大量增加及对痛刺激感受增强。
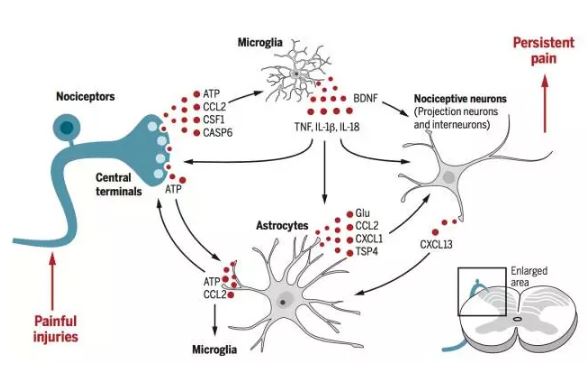
图3
目前在肿瘤和疼痛领域中的研究均是围绕着肿瘤怎样引起疼痛的机制进行,纪如荣教授则从另一个方面思考,即一些肿瘤早期具有无痛的表现,那么其具体机制又是什么样的呢?在西方国家中黑色素瘤的发病率较高,并且具有无痛的临床表现,而皮肤肿瘤位于体表也易于观察,是一个探讨肿瘤细胞与疼痛关系的理想模型。
PD-L1镇痛机制
PD-L1全称为程序性死亡受体-配体1,是由肿瘤细胞产生的一类跨膜蛋白,可与位于免疫细胞T细胞上分布的PD-1受体结合,传导抑制性下游信号,从而产生免疫抑制和免疫耐受,使肿瘤细胞得以逃避T细胞的摧毁。临床上采用PD-1抗体的免疫疗法,通过阻止PD-1和PD-L1的识别过程,部分恢复T细胞功能,使T细胞可以杀死肿瘤细胞。该疗法在黑色素瘤的临床试验中效果显著,提高了患者生存率。
那么肿瘤细胞分泌的PD-L1会不会与黑色素瘤的无痛临床表现有关呢?为了探讨这个问题,纪教授课题组建立了小鼠足底黑色素瘤的模型(图4),肿瘤注射在足底便于采用传统的多种疼痛检测方法进行测量。实验发现,随着造模时间延长,皮下的肿瘤显著增长,而小鼠患侧足底的自发痛与诱发痛却始终无明显变化,提示该肿瘤早期发展过程中癌痛可能被某些机制掩盖了。此外他们也观察到,肿瘤组织中PD-L1的含量远高于正常的皮肤和肌肉组织,肿瘤小鼠的血清PD-L1含量也显著高于假手术小鼠。PD-L1是否是造成癌痛被掩盖的主因吗?接下来他们尝试去阻断PD-L1和PD-1受体结合这个过程,使用了nivolumab这个已经在美国上市的抗PD-L1的药,发现小鼠的自发痛显著增加,同时in vivo记录的坐骨神经自发放电也显著增加。我们知道上市的药是根据人源的蛋白合成的抗体,因此他们换用小鼠特异的PD-1抗体重复了这一实验,仍得到了相似的结果。这说明干扰PD-L1和PD-1受体结合这个过程可以使黑色素瘤小鼠被掩盖的癌痛表露出来,同时提示PD-L1本身可能具有一定镇痛作用。
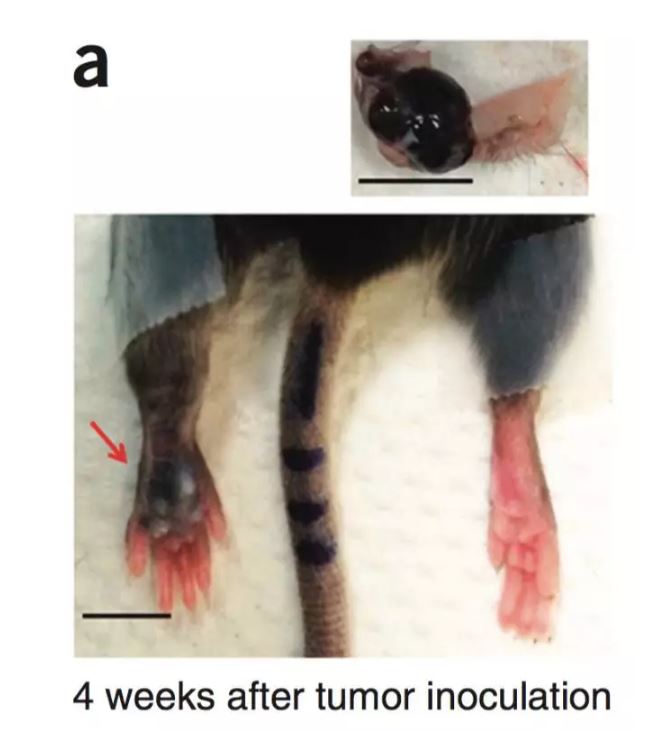
图4
进一步的研究显示,在其他疼痛模型中如福尔马林介导的急性炎症痛模型,PD-L1注射后可显著缓解第二相的自发痛反应。纪教授课题组通过ELISA的实验检测了小鼠各类组织中PD-L1的含量,发现正常的组织细胞也可分泌一定的PD-L1,其中在外周及中枢神经系统中均可检测到,提示PD-L1可能也是一个内源性的疼痛抑制分子。分子生物学实验显示,PD-L1的受体PD-1在野生型小鼠的DRG及外周神经纤维中也有广泛表达。为了进一步探讨PD-L1与PD-1的镇痛机制,纪教授课题组构建了PD-1敲除小鼠,并发现PD-1敲除小鼠的基础机械痛与热痛阈值均显著下调,即对痛刺激的敏感性增强。接下来在背根神经节DRG神经元上的电生理实验发现,PD-L1可以抑制动作电位的发放,降低静息膜电位,使细胞对外界刺激的反应性降低。在PD-1敲除小鼠则观察不到这样的现象,说明PD-L1是通过PD-1发挥其作用的。
PD-1受体发挥其抑制性功能依赖下游信号SHP-1的磷酸化激活。后续实验结果显示,小鼠给予PD-L1后,DRG中pSHP-1的表达显著上调;而对小鼠同时给予PD-L1和SHP-1的抑制剂SSG后,PD-L1的镇痛效果消失,说明PD-L1的镇痛作用依赖于PD-1下游SHP-1的激活。电生理实验显示,PD-L1可以抑制DRG神经元的钠电流,同时可以激活负责维持静息膜电位的钾通道TREK2,从而达到抑制动作电位的发放和降低静息膜电位的效果。
上述实验围绕着PD-L1在外周镇痛的机制进行,前面也提到了PD-L1在外周及中枢均可被检测到,那么PD-L1是否可能在中枢作为一个神经调质也发挥镇痛作用呢?后续研究发现,鞘内注射的方式给予PD-L1,可以对神经病理痛和骨癌痛产生显著的镇痛效果,并且通过电生理实验证实,其机制是PD-L1可抑制脊髓神经元的突触传递、降低WDR神经元(脊髓背角广动力型神经元,被证实与慢性痛的中枢敏化密切相关)的活性。
除此之外,纪教授课题组还完成了在人DRG神经元上的实验,证实了PD-L1与PD-1受体相互作用的结果与小鼠类似。电生理实验显示,在离体培养的人的小直径DRG神经元上给予PD-L1,也可以观察到动作电位发放的减少和静息膜电位下调的结果。
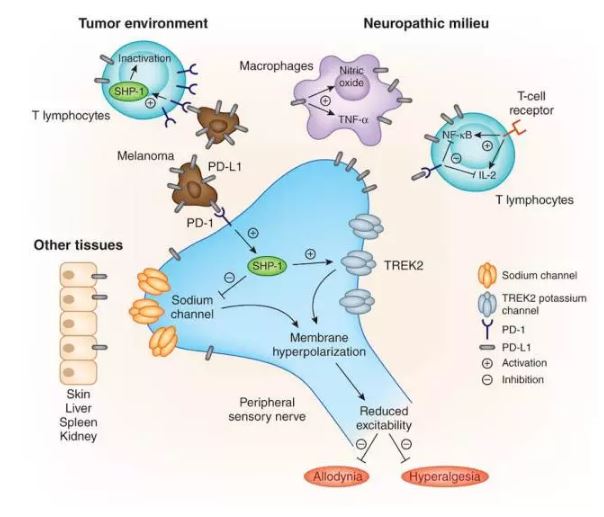
图5
纪如荣教授总结认为该研究主要阐述了以下几个新观点:1)PD-L1是一个内源性的疼痛抑制剂;2)PD-L1是一个广泛表达在外周及中枢神经系统的神经调质;3)PD-L1参与掩盖了黑色素瘤的癌痛表现;4)提供了一个观察疼痛与免疫治疗的新角度。纪教授认为这一研究对临床具有一定的提示意义,可以进一步观察接受治疗的患者中是否存在产生癌痛的情况,也可以通过对患者的患处痛阈进行检测,初步、快速判断是否适合PD-L1免疫疗法等。讲座最后,在场的老师和同学就PD-L1的镇痛机制、PD-L1疗法的临床观察、黑色素瘤小鼠模型中PD-L1的抑癌效果及测痛方式等进行了深入的讨论。本次学术报告在热烈的氛围中圆满结束。
参考文献
1. Chen G, Kim YH, Li H, Luo H, Liu DL, Zhang ZJ, Lay M, Chang W, Zhang YQ, Ji RR (2017) PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nature Neuroscience 20:917-926.
2. Hirth M, Gandla J, Kuner R (2017) A checkpoint to pain. Nature neuroscience 20:897-899.
3. Ji RR, Xu ZZ, Gao YJ (2014) Emerging targets in neuroinflammation-driven chronic pain. Nature Reviews Drug Discovery 13:533-548.
4. Ji RR, Chamessian A, Zhang YQ (2016) Pain regulation by non-neuronal cells and inflammation. Science (New York, NY) 354:572-577.