陈家明:Immunogenic Cell Death: The Good, the Bad and the Ugly
3月23日,北京大学IDG麦戈文脑科学研究所邀请浙江大学陈家明教授,在金光楼邓祐才报告厅作了精彩的学术报告,标题为“Immunogenic Cell Death: The Good, the Bad and the Ugly”。陈良怡教授主持报告会。本期学术笔记由该报告整理而成。
陈家明教授探讨了坏死性凋亡的信号通路,以及它在组织复修,和各类炎症疾病中的功能。
TNF(肿瘤坏死因子)能够抑制肿瘤的生长(图1)。与此同时使用TNF治疗的病人也会变得十分消瘦。TNF会激活NF-κB 可以触发了另一种形式的细胞死亡也就是坏死性凋亡(necroptosis)。坏死性凋亡是细胞死亡的一种溶解性和炎症性形式。
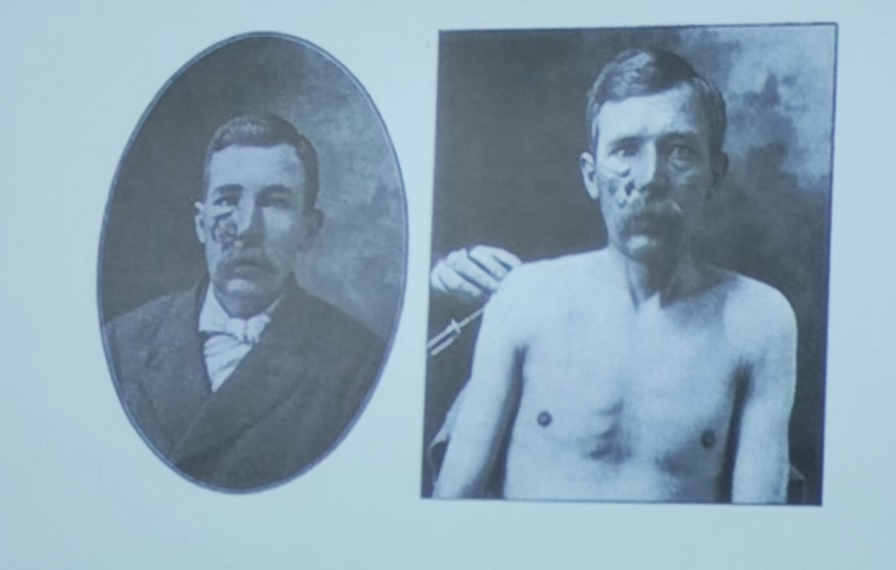
图1. 病人在使用TNF治疗之后(右侧)面部肿瘤生长受到抑制。
细胞坏死需要RIPK1-RIPK3-MKL轴的参与(如图2C)。RIPK1包含两个重要的结构N-端激酶结构域以及RIP同型相互作用模块 (RHIM)。RIPK1和RIPK3之间通过RHIM的同型相互作用形成坏死小体复合物,从而通过磷酸化级联激活MLKL。磷酸化MLKL发生低聚并迁移到质膜,在那里它通过引发膜破裂来诱导坏死。一般情况下不会发生细胞坏死。但当caspase8被抑制时,才会激活RIPK1和RIP3引发的坏死性凋亡过程[1]。
除此之外, RIPK3/MKL依赖性的坏死性凋亡能够被TLRs(Toll-like receptors,TLR)或者病毒激活。给小鼠注射病毒会诱导TNF的产生(图3B),RIPK1-RIPK3的产生(图3A)以及组织的细胞坏死(图3C)。RIP3缺陷小鼠表现出严重受损的病毒诱导的组织坏死、炎症和病毒复制控。因此,RIP3 通过启动促坏死激酶级联控制程序性坏死,该级联对于针对病毒感染的先天炎症反应至关重要[2]。
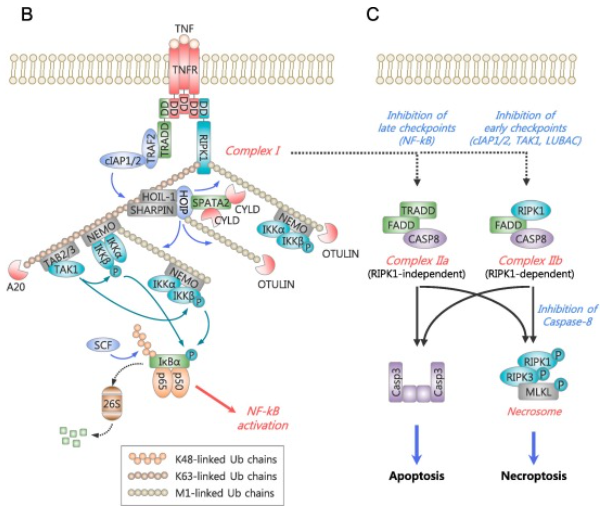
图2. TNF可诱发坏死性凋亡
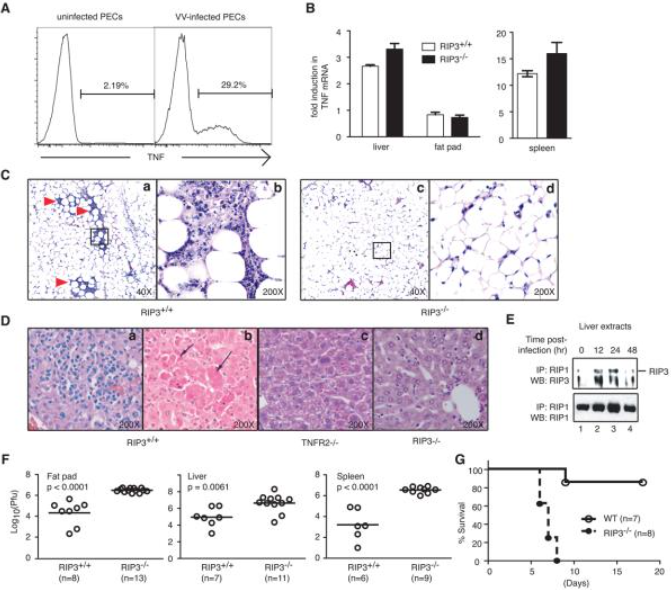
图3. RIP3依赖性程序性坏死是控制痘苗病毒在体内复制所必需的
B:病毒感染野生型和RIP3敲除小鼠。感染后24小时在腹膜渗出细胞中检测到TNF表达。
C:在野生型小鼠中,VV感染导致内脏脂肪垫中以中性粒细胞/巨噬细胞浸润为标志的炎症(图3C,红色箭头),而RIP3敲除小鼠中明显不存在这种炎症
E: 病毒感染的肝脏中RIP1-RIP3复合物的形成。
F: 在感染后第3天测定RIP3野生型和RIP3敲除鼠内脏脂肪垫、肝脏和脾脏中的病毒滴度。
G:VV感染RIP3野生型和RIP3敲除鼠的生存图。
牛痘病毒是一种非常古老的病毒,有高度的免疫原性,但会引起相对良性的疾病,曾被用来大范围地接种。陈家明团队经检测发现,牛痘病毒(CPXV)对TBZ诱导的坏死具有耐受性(图4)这种耐性是由于RIPK3的降解导致的(图5)。为了进一步探究背后的机制,陈家明团队对牛痘病毒基因进行集中的siRNA筛选,筛选出了CPXV006基因,它编码一种在N末端有六个锚蛋白重复序列和一个C末端F-Box的蛋白质(图6A)。该基因被命名为vIRD。删除vIRD完全阻止了CPXV诱导的RIPK3降解(图6B)。感染CPXV-ΔvIRD的L929细胞发生自分泌TNF诱导的坏死(图6C)。随后作者证明vIRD诱导的RIPK3降解需要依赖SCF(SKP1-Cullin1-Fbox)复合物
哺乳动物F-box蛋白作为衔接蛋白,将蛋白质底物募集到SKP1-Cullin1-Fbox(SCF)复合物中,这是一种促进蛋白质泛素化和蛋白酶体降解的多亚基复合物[3]。CPXV感染诱导了vIRD、SKP1和CUL1之间的特异性相互作用(图6E)。
siRNA对CUL1的沉默逆转了CPXV诱导的RIPK3降解,并恢复了对自分泌TNF诱导的坏死的敏感性(图6F-G)。SCF活性由CUL1烯二基化激活[4]。抑制剂MLN4924有 效地将所有细胞CUL1转化为快速迁移的非活性形式,逆转了CPXV诱导的RIPK3降解,并恢复了CPXV诱发的L929细胞对坏死的敏感性(图6H)。在MLN4924存在的情况下,抗TNF中和抗体部分抑制了CPXV介导的细胞死亡。这些结果表明:vIRD以SCF依赖的方式靶向RIPK3降解以抑制TNF、RIPK1和RIPK3诱导的坏死[5]。
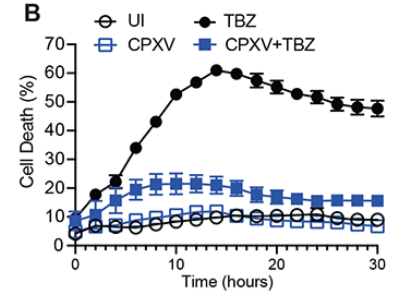
图4. 牛痘病毒感染细胞对坏死凋亡具有抗性(TBZ处理会诱导典型的坏死性凋亡)
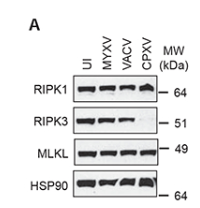
图5. vIRD触发了RIPK3蛋白的降解
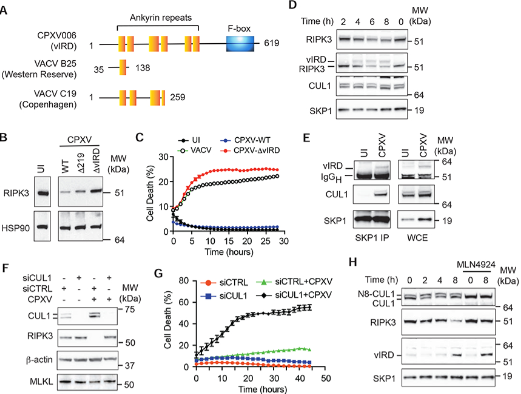
图6. vIRD与细胞SCF复合物组分相互作用,以促进RIPK3降解和坏死抗性
(A)来自CPXV的vIRD的结构域组织示意图以及来自VACV Western Reserve和Copenhagen菌株的截短vIRD直向同源物。
(B) 通过蛋白质印迹分析用WT或指示的突变体CPXV感染的L929细胞的RIPK3表达。
(C) vIRD对于感染CPXV的细胞的坏死抵抗是必不可少的。使用Incucyte监测感染VACV、vIRD缺失的CPXV(CPXV-ΔvIRD)或vIRD回复体(CPXV-WT)的L929细胞的细胞死亡。结果为平均值±SEM。
(D) 在CPXV感染后的指定时间,通过蛋白质印迹测定vIRD表达和RIPK3降解的动力学。
(E) 病毒SCF复合物的形成。SKP1是从CPXV感染的和未感染的L929细胞中免疫沉淀的。通过蛋白质印迹测定CPXV感染后的vIRD和CUL1募集。
(F) CUL1对CPXV诱导的RIPK3降解至关重要。通过蛋白质印迹监测用CUL1特异性或对照扰乱的siRNA转染的L929细胞的RIPK3降解。
(G) CUL1对于CPXV介导的对坏死的抵抗是必不可少的。通过Incucyte活细胞成像监测用指示的siRNA转染的L929细胞的细胞死亡。结果为平均值±SEM。
(H) CUL1 nedylation对CPXV诱导的RIPK3降解的影响。用MLN4924处理未感染或CPXV感染的L929细胞。通过蛋白质印迹法测定vIRD和RIPK3的表达。
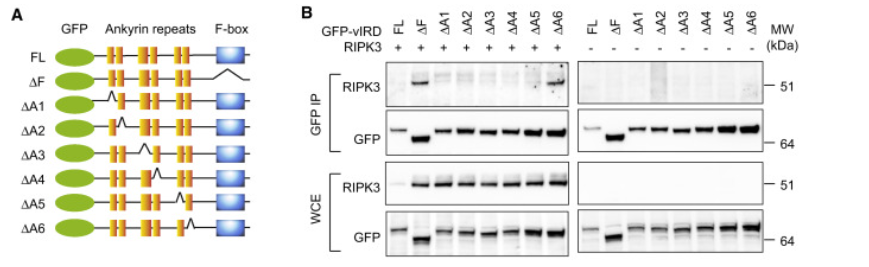
图7. vIRD的锚蛋白重复序列介导与RIPK3的结合
(A) 使用的vIRD突变体的示意图。
(B) 用指示的GFP标记的vIRD和RIPK3转染HEK293T细胞。使用所指示的抗体进行免疫沉淀和蛋白质印迹。
以上,陈家明团队从牛痘病毒中筛选到一个可以降解RIPK3的分子,称为vIRD。vIRD基因通过ankyrin结构域结合RIPK3,通过F-Box 结构域与SKP1和Cullin1形成SCF(SKP1-Cullin1-Fbox)复合物介导了RIPK3的降解,从而对细胞坏死产生耐性(图8)。
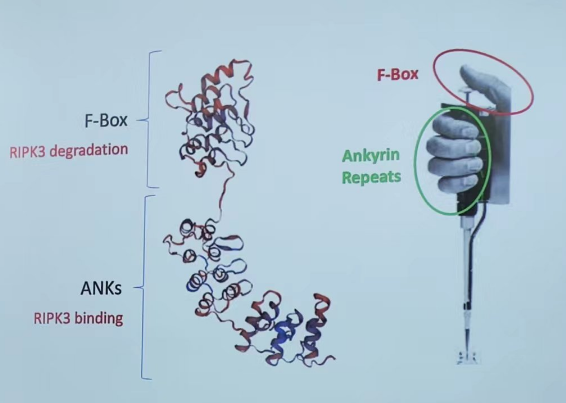
图8. vIRD介导细胞坏死抗性重要结构域
除此之外,陈教授团队发现细胞坏死性凋亡能够免疫保护小鼠应对肿瘤挑战。并且RIPK3在诱导细胞死亡的同时,也在组织再生中发挥关键作用。当树突细胞Ripk3失活小鼠受到DSS处理诱发结肠炎时,它们表现出比同窝出生的野生型小鼠更严重的体重减轻和结肠缩短(图9B–C)。树突细胞Ripk3失活小鼠在DSS治疗后的结肠中产生的IL-22(组织修复的关键启动子)比同窝对照显著减少(图9D)[6]。
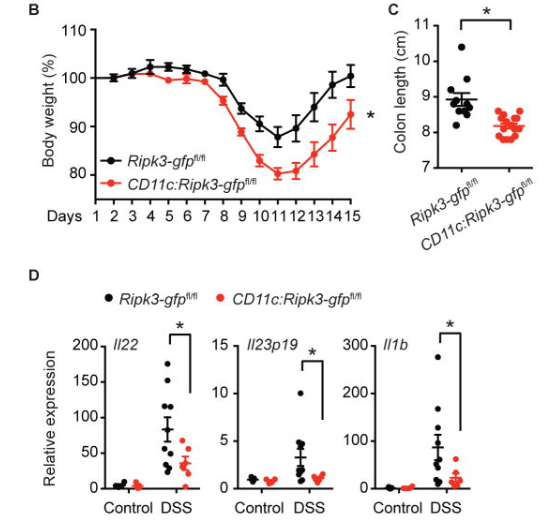
图9. 树突细胞特异性缺失RIPK3 RHIM对DSS诱导的大肠炎具有保护作用
B: DSS处理小鼠体重示意图。Ripk3-gfpfl/fl表示携带GFP报告基因的小鼠;CD11c:Ripk3-gfpfl/fl表示小鼠Ripk3 基因RHIM domain失活。
C:DSS处理小鼠结肠长度示意图。
D:组织修复关健启动子表达水平示意图。
撰文:牛梦晓
审核:陈良怡
参考文献
[1] Roberts JZ, Crawford N, Longley DB. The role of Ubiquitination in Apoptosis and Necroptosis. Cell Death Differ. 2022 Feb;29(2):272-284. doi: 10.1038/s41418-021-00922-9. Epub 2021 Dec 15. PMID: 34912054; PMCID: PMC8817035.
[2]Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009 Jun 12;137(6):1112-23. doi: 10.1016/j.cell.2009.05.037. PMID: 19524513; PMCID: PMC2727676。
[3]Lee EK, Diehl JA. SCFs in the new millennium. Oncogene. 2014 Apr 17;33(16):2011-8. doi: 10.1038/onc.2013.144. Epub 2013 Apr 29. PMID: 23624913.
[4]Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015 Jan;16(1):30-44. doi: 10.1038/nrm3919. PMID: 25531226; PMCID: PMC5131867.
[5]Liu Z, Nailwal H, Rector J, Rahman MM, Sam R, McFadden G, Chan FK. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity. 2021 Feb 9;54(2):247-258.e7. doi: 10.1016/j.immuni.2020.11.020. Epub 2021 Jan 13. PMID: 33444549; PMCID: PMC7878414.SCF
[6] Moriwaki K, Balaji S, Bertin J, Gough PJ, Chan FK. Distinct Kinase-Independent Role of RIPK3 in CD11c+ Mononuclear Phagocytes in Cytokine-Induced Tissue Repair. Cell Rep. 2017 Mar 7;18(10):2441-2451. doi: 10.1016/j.celrep.2017.02.015. PMID: 28273458; PMCID: PMC5343671